Asymmetric Catalysis
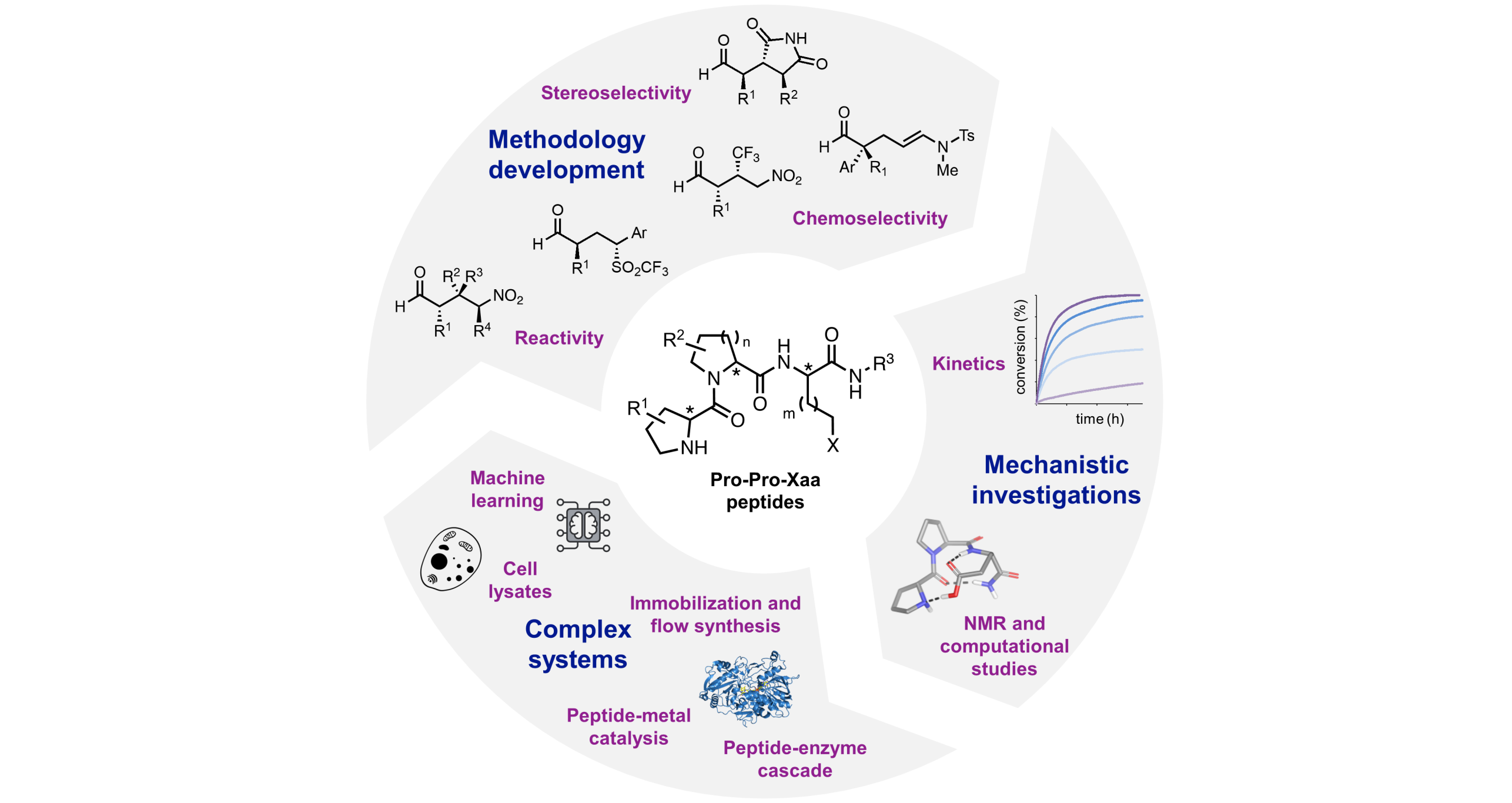
Asymmetric Catalysis with Peptides
In nature, peptides and other small molecules fulfill manifold different functions, but not a single natural peptide with catalytic activity is known. Instead, catalysis in nature is a hallmark of enzymes. We are intrigued by the question of whether short peptides can function as catalysts. This research established effective stereoselective catalysts for reactions that are of great interest, e.g., for the production of pharmaceuticals, and tackles the question of whether peptides have played a role in the chemical evolution of enzymes.
With a combination of combinatorial screening and rational design we established tripeptides of the general type H-Pro-Pro-Xaa as effective catalysts for C–C bond formations. Their modular nature allowed us to identify stereoselective catalysts for even highly substituted substrates that are otherwise reluctant to engage in addition reactions with exquisite yields and stereoselectivities. The peptide catalysts are so reactive that many of the reactions proceed at a catalyst loading that is lower than that needed with other chiral amines. The peptides are also so robust that an immobilized peptide catalyst can be used in a flow reactor for the continuous synthesis of more than 100 g of product without loss in activity and stereoselectivity. The chemoselectivity of some of our peptide catalysts is so high that stereoselective one-pot cascade reactions with enzymes and reactions in cell lysates are feasible. Our mechanistic investigations with a combination of kinetic, NMR-spectroscopic, and ESI-MS studies rationalized the exquisite performance of the peptidic catalysts and provided general guidelines for the design of stereoselective catalysts, beyond peptide catalysis.
We are currently extending our studies to other challenging and relevant reactions and explore a potential role of peptide catalysts in prebiotic chemistry.
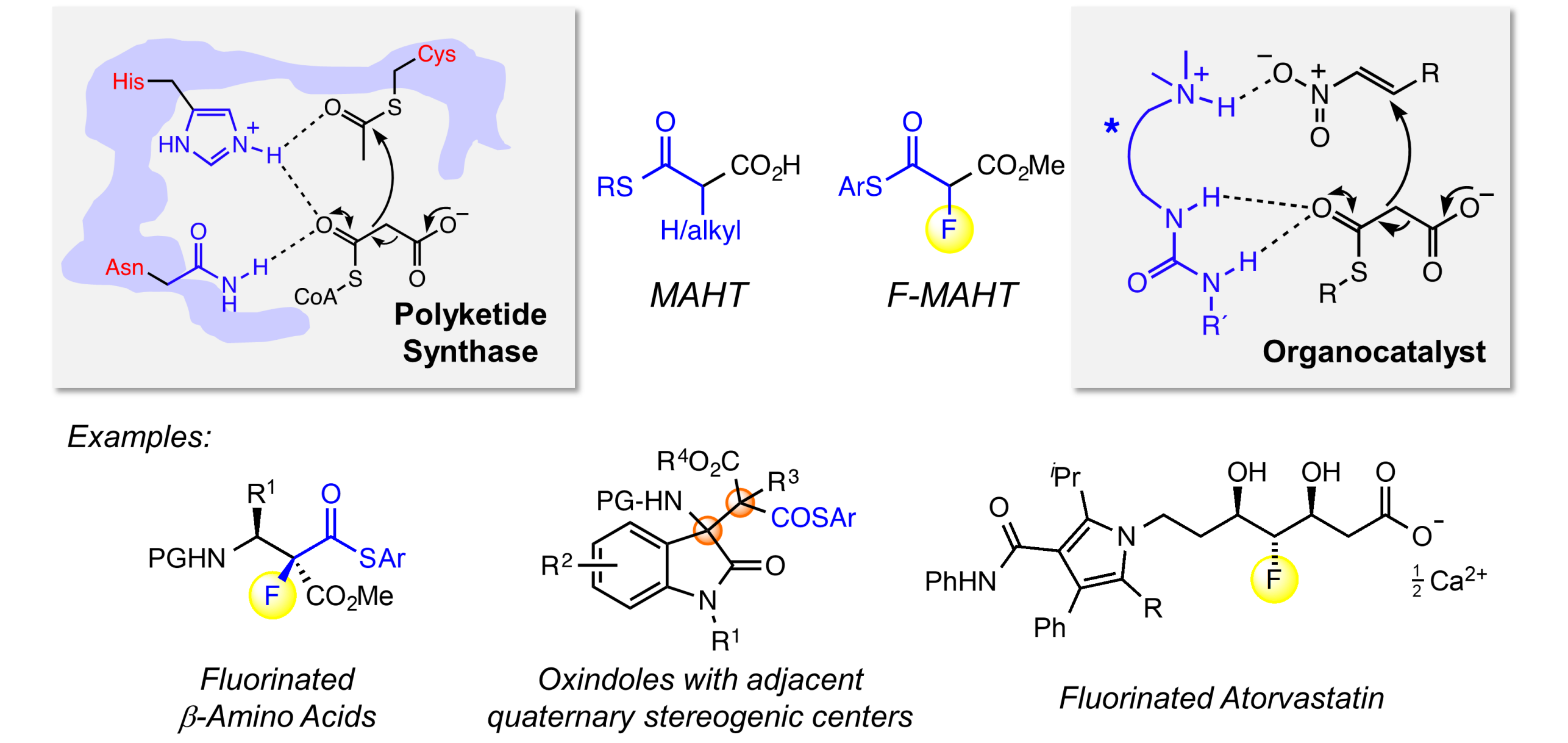
Polyketide Synthase-inspired Organocatalysis
Inspired by natural polyketide synthases, which use malonic acid half thioesters (MAHTs) as thioester enolate equivalents, we developed organocatalytic methods for stereoselective addition reactions of MAHTs and protected variants (monothiomalonates, MTMs) to electrophiles. Addition products include synthetically versatile compounds bearing, e.g., 4° stereogenic centers and two adjacent fully substituted centers as well as β-aminothioesters that allow for coupling reagent-free solid phase peptide synthesis. We employ hydrogen-bond donor catalysts build on a chiral scaffold for this type of transformation.
Addition reactions with fluorinated MAHTs enabled, for the first time, enantioselective fluoroacetate aldol reactions, a transformation that had so far remained elusive and provides access to fluorinated analogues of medicinally relevant acetate-derived compounds, such as statins. We are now using these organocatalytic transformations for catalytic length-controlled oligomerizations and the synthesis of fluorinated polyketides.
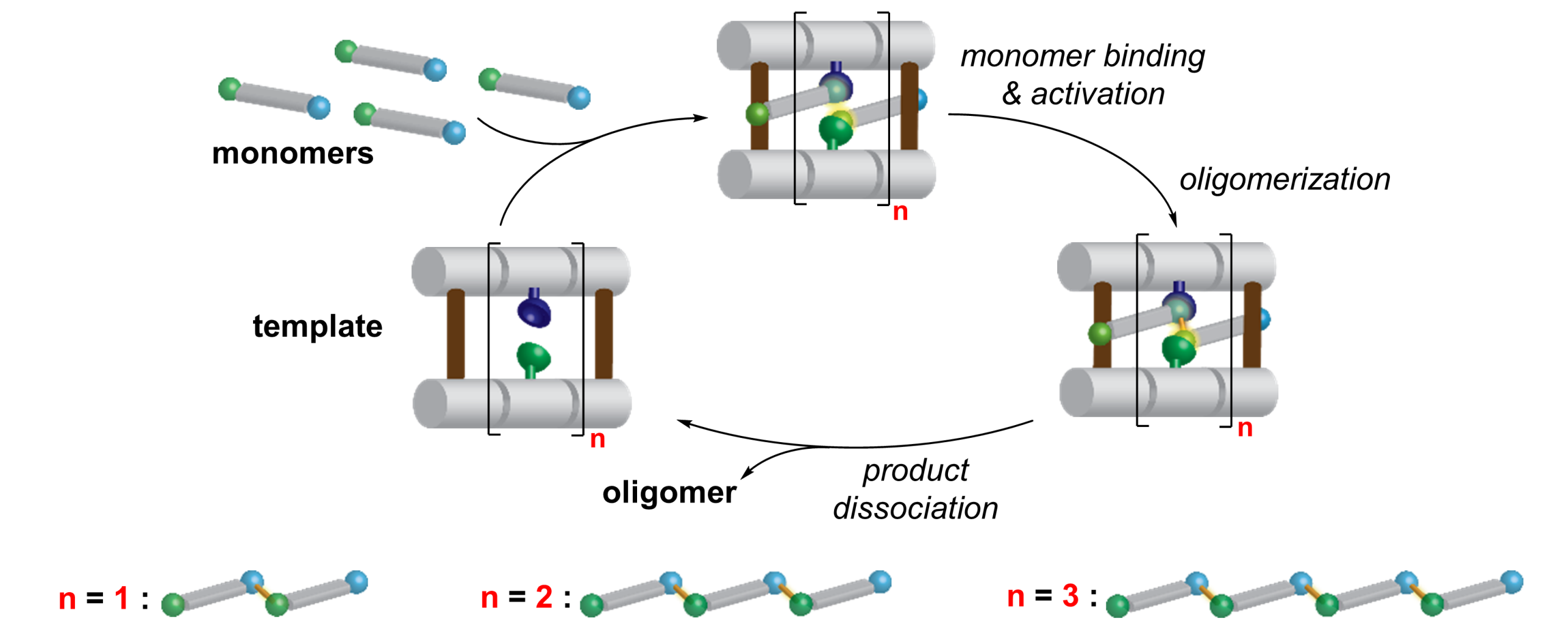
Catalytic Templated Oligomerization
Nature uses templated synthesis to produce oligomers from monomer building blocks. Templated synthesis is also an intriguing strategy for the synthesis of oligomers in the laboratory since it avoids labor-intense stepwise syntheses. A common limitation of templated oligomerization is the tight binding between the template and the oligomer. The newly formed oligomer is, therefore, only accessible in stoichiometric amounts relative to the template, and the complex between the template and the synthetic oligomer needs to be disassembled in a subsequent release step.
We achieved the first catalytic length-controlled oligomerization. Macrocyclic templates catalyzed the formation of oligomers through aldol-type reactions of a bifunctional substrate bearing aldehyde and malonic half thioester moieties. Two rigid oligoproline moieties position urea and amino groups for substrate binding and activation at distances related to the distance between the reactive sites within the substrate. The reaction converts a 1,3-dicarbonyl moiety with two sp2-carbons into a ß-hydroxy thioester with an sp3-carbon and concomitant release of CO2. As a result, the template binds the oligomer product less tightly than the monomers, thus, allowing for product release and catalytic turnover.